
Utilization of membrane vesicle preparations to study drug-ABC-transporter interactions
Glavinas H, Méhn D, Jani M, Oosterhuis B, Herédi-Szabó K, Krajcsi P Solvo Biotechnology Contents 1. Abstract Introduction 2. Retrospective 3. Assay types 4. Membrane preparations 5. Correlations of membrane test data with data from other in vitro and in vivo methods 6. Conclusion 7. Expert opinion 8. Annotated bibliographyAbstract
Background: The last 15 years marked an expansion in our understanding of how ABC transporters modulate the pharmacokinetic properties of drugs. Assays based on different membrane preparations were one of the first methods developed to study ABC transporters. Later, they turned out to be valuable tools to gain insight into the nature of drug-ABC transporter interactions. Objectives: Membranes prepared from different sources have been used and characterized; based on the biochemical characteristics of the transport process a number of different assay types have been developed. Methods: This review focuses on the current experiences on how different membrane-based assays can be utilized in pharmaceutical R&D. Sources of membrane preparations, available assay types and correlation studies between different in vitro and in vivo methods are discussed. Results/conclusion: Membrane based assays are valuable tools in drug discovery to characterize drug-ABC transporter interactions. Keywords: ABC transporter, ATPase assay, vesicular transport assay, nucleotide trapping assay, monolayer assayIntroduction
ABC proteins form one of the largest protein families known. The family is characterized by a conserved structure of ATP binding domains (containing the "ATP Binding Cassette" motif) and transmembrane domains. In mammals, the functional ABC protein contains two ATP binding domains and two transmembrane domains. The four domains can be present in one polypeptide chain ("full transporters") or might be set up by the homo- or heterodimerization of two polypeptides containing one of each domain ("half transporters"). Each transmembrane domain forms 6 transmembrane alpha-helices spanning the membrane. The two transmembrane domains are believed to form a pore-like structure creating a channel across the membrane. The transmembrane domains are thought to exist in an "open" and "closed" conformation regulated by ATP binding and hydrolysis. The change in conformation is thought to trigger the translocation of compounds across the cell membrane, might open and close ion channels (e.g. ABCC7) or serve as regulators for complex, multi-protein ion channels (e.g. ABCC8, ABCC9). Homology searches using the conserved ABC motif revealed 48 ABC proteins in the human genome. These proteins are grouped into 7 families based on gene structure, order of the domains, and sequence homology. The families are designated with letters A to G, and the following number refers to the specific transporter (reviewed in [1]). ABC transporters transport substrates across the cell membrane against their concentration gradient (active transport). The energy requirement for this process is derived from ATP hydrolysis. It is believed that ATP binding and hydrolysis generates a conformation change in the ATP binding domain. The close interaction between the ATP binding domains and the transmembrane domains creates the transmission of force to the transmembrane domains resulting in a conformation change in the transmembrane domains driving substrate translocation. Several members of the ABC protein family were shown to be active transporters (reviewed in [2]). The last 15-20 years extensive research in this area resulted in a significant advance in our understanding of how different ABC transporters modulate the different pharmacokinetic parameters of xenobiotics (reviewed in [3], [4]). It turned out that multiple ABC transporters located at different pharmacological barriers are important determinants of ADME (Absorption-Distribution-Metabolism-Excretion) properties of drug molecules (table 1.). The discovery of the importance of ABC transporter interactions in the ADME properties of drugs drew the attention of the pharmaceutical industry. Scientists involved in drug development realized that by examining the interactions of the transporters with pharmacologically active agents the cellular and tissue distribution of these compounds can be predicted. Transporter interactions have a major impact on the later phases of drug development, therefore, in accordance with the "fail fast - fail cheap" paradigm, pharmaceutical industry requires low-cost, high-throughput methods for early testing. Although knock-out mice, as well as mutant mice and rat strains deficient in the expression of functional ABC transporter proteins are available these techniques are not suitable for high throughput studies required in the early ADME phase of drug discovery. In vitro ABC transporter assays rely on two basic principles: (1) use whole cells expressing the transporter(s) or (2) utilize membrane preparations containing the transporter(s) studied (reviewed in [1]). This review is focusing on the membrane based assays used in ABC transporter research. The current understanding on which membranes and which assay types match the different needs of pharmaceutical research and development is reviewed.| Standard name | Other abbreviations | Definition |
| ABCB1 | MDR1/P-gp | Multidrug resistance protein/P-glycoprotein |
| ABCB11 | BSEP/sPGP | Bile salt export pump/sister of P-glycoprotein |
| ABCC1 | MRP1 | Multidrug resistance associated protein 1 |
| ABCC2 | MRP2/cMOAT | Multidrug resistance associated protein 2 |
| ABCC3 | MRP3 | Multidrug resistance associated protein 3 |
| ABCC4 | MRP4 | Multidrug resistance associated protein 4 |
| ABCG2 | BCRP/MXR | Breast cancer resistance protein |
Retrospective
Membrane preparations have been used to study transport processes before the discovery of ABC transporters. One of the best known early examples are the calcium uptake studies conducted on muscle microsome preparations, where ATP dependent uptake of calcium was detected in membranes prepared from the sarcoplasmic reticulum of these cells [5].In 1970 Steck and coworkers showed that inside-out plasma membranes can be purified from human red cells using low ionic strength and divalent cations. The inside-out vesicles can be separated from right-side-out vesicles by centrifugation to equilibrium in dextran density gradients [6], [7]. In 1976 the first ABC transporter involved in drug transport (P-glycoprotein, multidrug resistant transporter 1 (MDR1), or also known as ABCB1) was identified [8] and later cloned ([9], [10], [11]). Applying inside out membrane vesicles prepared from cells expressing ABCB1 the ATP dependent vesicular transport of vinblastine [12] and colchicin [13] was demonstrated. The same year membranes purified from Sf9 insect cells expressing ABCB1 were used to show that the ATPase activity of the transporter is stimulated in the presence of ABCB1 substrates [14]. In 1995 the nucleotide trapping assay for ABCB1 was described using plasma membranes from a multidrug-resistant Chinese hamster ovary cell line [15]. These membrane based methods have been refined, further developed since and are currently widely used in pharmaceutical research and development to detect the interaction of drugs and ABC transporters.
Assay types
Membrane preparations from various sources have been utilized in different biochemical assays [16]. The three most commonly used membrane assays are the ATPase, the vesicular transport and the nucleotide traping assay. The first relies on the changes in the hydrolysis rate of ATP in the presence of interacting compounds, the second method detects the translocation of substrates into inside-out vesicles and the latter measures the occlusion of the nucleotide during the transport catalytic cycle. A short overview on the principle of these assays is given and technical issues are highlighted with examples.
| A. | B. |
 | |
 Figure 2. Typical results of direct (A) and indirect (B) vesicular transport experiments. The assays were performed using SB-BSEP-Sf9-VT membrane vesicles (Solvo Biotechnology, Budaörs, Hungary) according to a protocol based on Noe et al. [70]. The ATP dependent transport of taurocholate is saturable indicating that a carrier is involved in the process. Compounds that interact with the transporter (e.g. cyclosporine A) inhibit the ATP dependent taurocholate transport.
3.2 The ATPase assay
ABC transporters use ATP as energy source to transport substrates across cell membranes. The ATPase assay is based on the principle that compounds interacting with the transporter modulate ATPase activity of the latter (figure 1B). The ATPase activity can be detected as the amount of inorganic phosphate released by the enzyme [14]. This assay is most commonly used for ABCB1 (MDR1, Pgp) and for ABCG2 (BCRP, MXR) studies, although it is also available for different MRPs ([30], [31], [32], [33]).
It is generally accepted that ATP cleavage is tightly linked to substrate translocation as uptake of substrates into membrane vesicles is an ATP dependent process [13]. Mutations in the ATPase domain result in loss of transport function [34] and the activity of the multidrug transporter in drug-resistant cells is associated with rapid cellular ATP depletion when ATP synthesis is inhibited [35]. Most substrates when tested at multiple concentrations show a "bell shaped" curve: at lower concentrations the ATPase activities increase while at higher concentrations it decreases. The currently accepted hypothesis for this phenomenon is a two binding site model. The transporter harbors two distinct binding sites for the substrate molecule: one with high affinity, activating / transport site localized in the cytoplasmic or transmembrane domains and a second, low-affinity, inhibitory site localized most likely in the extracellular domains, often termed as "release site" ([36], [37], [38]). The relative affinity of the two binding sites is different for the different substrates resulting in different ATPase curves. If the affinities of the two binding sites are very different (i.e. the affinity of the inhibitory site is orders of magnitude lower than the affinity of the activating site) Michaelis-Menten type curves are observed (figure 3A) while, if the binding constants are close to each other the observed curve is bell-shaped (figure 3B). It is therefore important to measure interaction of drugs in the ATPase assay at multiple concentrations in a wide range otherwise interactions could be "missed". The "bell shaped" nature of ATPase curves might also arise from substrate-interference with the color reaction at high concentrations of the test compound [39].
Some compounds are translocated very slowly by the transporter which results in a slow rate of ATP hydrolysis that do not yield detectable amount of inorganic phosphate. Typical example for such interactions is the ABCB1 mediated transport of cyclosporine A. Although cyclosporine A was shown to be transported by ABCB1 [40], no stimulation of the transporter ATPase activity can be detected in the ATPase assay. To reduce the number of false nagatives the inhibition mode of the ATPase assay should be performed. In this case the modulation of the maximal transporter ATPase activity of the transporter triggered by a high turnover substrate is measured. Slowly transported compounds will compete with substrates transported with high turnover rate thereby inhibiting the transporter ATPase activity. This assay will not distinguish between slowly transported substrates and inhibitors (e.g. cyclosporin A and GF120918, respectively, figure 3C-D); however, it will indicate the interaction for slowly transported substrates that are not detected in the ATPase activation assay.
The ATPase assay is one of the most widely used screening tools for ABCB1 in the pharmaceutical industry. It is a simple and reproducible assay that detects most of the transporter-drug interactions when performed in both in the activation as well as inhibition modes. The ATPase assay is now also available for other efflux transporters and will probably be an important screening tool for transporters that are involved in clinically relevant drug-transporter interactions.
Figure 2. Typical results of direct (A) and indirect (B) vesicular transport experiments. The assays were performed using SB-BSEP-Sf9-VT membrane vesicles (Solvo Biotechnology, Budaörs, Hungary) according to a protocol based on Noe et al. [70]. The ATP dependent transport of taurocholate is saturable indicating that a carrier is involved in the process. Compounds that interact with the transporter (e.g. cyclosporine A) inhibit the ATP dependent taurocholate transport.
3.2 The ATPase assay
ABC transporters use ATP as energy source to transport substrates across cell membranes. The ATPase assay is based on the principle that compounds interacting with the transporter modulate ATPase activity of the latter (figure 1B). The ATPase activity can be detected as the amount of inorganic phosphate released by the enzyme [14]. This assay is most commonly used for ABCB1 (MDR1, Pgp) and for ABCG2 (BCRP, MXR) studies, although it is also available for different MRPs ([30], [31], [32], [33]).
It is generally accepted that ATP cleavage is tightly linked to substrate translocation as uptake of substrates into membrane vesicles is an ATP dependent process [13]. Mutations in the ATPase domain result in loss of transport function [34] and the activity of the multidrug transporter in drug-resistant cells is associated with rapid cellular ATP depletion when ATP synthesis is inhibited [35]. Most substrates when tested at multiple concentrations show a "bell shaped" curve: at lower concentrations the ATPase activities increase while at higher concentrations it decreases. The currently accepted hypothesis for this phenomenon is a two binding site model. The transporter harbors two distinct binding sites for the substrate molecule: one with high affinity, activating / transport site localized in the cytoplasmic or transmembrane domains and a second, low-affinity, inhibitory site localized most likely in the extracellular domains, often termed as "release site" ([36], [37], [38]). The relative affinity of the two binding sites is different for the different substrates resulting in different ATPase curves. If the affinities of the two binding sites are very different (i.e. the affinity of the inhibitory site is orders of magnitude lower than the affinity of the activating site) Michaelis-Menten type curves are observed (figure 3A) while, if the binding constants are close to each other the observed curve is bell-shaped (figure 3B). It is therefore important to measure interaction of drugs in the ATPase assay at multiple concentrations in a wide range otherwise interactions could be "missed". The "bell shaped" nature of ATPase curves might also arise from substrate-interference with the color reaction at high concentrations of the test compound [39].
Some compounds are translocated very slowly by the transporter which results in a slow rate of ATP hydrolysis that do not yield detectable amount of inorganic phosphate. Typical example for such interactions is the ABCB1 mediated transport of cyclosporine A. Although cyclosporine A was shown to be transported by ABCB1 [40], no stimulation of the transporter ATPase activity can be detected in the ATPase assay. To reduce the number of false nagatives the inhibition mode of the ATPase assay should be performed. In this case the modulation of the maximal transporter ATPase activity of the transporter triggered by a high turnover substrate is measured. Slowly transported compounds will compete with substrates transported with high turnover rate thereby inhibiting the transporter ATPase activity. This assay will not distinguish between slowly transported substrates and inhibitors (e.g. cyclosporin A and GF120918, respectively, figure 3C-D); however, it will indicate the interaction for slowly transported substrates that are not detected in the ATPase activation assay.
The ATPase assay is one of the most widely used screening tools for ABCB1 in the pharmaceutical industry. It is a simple and reproducible assay that detects most of the transporter-drug interactions when performed in both in the activation as well as inhibition modes. The ATPase assay is now also available for other efflux transporters and will probably be an important screening tool for transporters that are involved in clinically relevant drug-transporter interactions.
 Figure 3. Typical ATPase curves: Michaelis-Menten (A), "bell shaped" (B), slowly transported compound (C), inhibitor (D). The assays were performed using SB-MDR1-Sf9-ATPase membrane vesicles (Solvo Biotechnology, Budaörs, Hungary) according to a protocol based on Sarkadi et al. . Some substrates of ABC transporters stimulate the vanadate sensitive ATPase activity of membrane preparations in a standard Michaelis-Menten way (A). Other substrates are stimulators at lower concentrations, while act as inhibitors at higher concentrations resulting in a "bell shaped" curve (B). It is believed that the inhibition at higher concentrations is due to a secondary, lower affinity binding site in the extracellular part of the transporter [36]. Slowly transported compounds (C) and inhibitors (D) do not stimulate the baseline vanadate sensitive ATPase activity, but inhibit the stimulated ATPase activity measured in the presence of a strong substrate. These two types of interactions cannot be distinguished using the ATPase assays.
3.3 The nucleotide trapping assay
For ABC transporters the catalytic cycle of transport and ATP hydrolysis involves the formation of a transition state complex. This complex contains the occluded nucleotide in the nucleotide binding site, which can be stabilized with vanadate, fluoroaluminate and beryllium fluoride (with the nucleotide ADP occluded in the binding pocket). This provides the theoretical basis for a screening method, called nucleotide trapping. Vanadate is the most studied trapping agent. It has been shown for a few transporters that the amount of nucleotide trapped in the binding site by vanadate is reflective of the rate of transport, so there is an increased binding in the presence of substrates [15]. Experimentally, the isolated cell membranes are incubated with alpha-32P labeled azido-ATP for a short time (30 seconds) and after washings in the presence of cold ATP the trapped, labeled nucleotides are covalently bound to the transporter by UV photo-cross linking. The bound nucleotide is visualized by phosphorimager after the separation of proteins with SDS-PAGE [41]. Though not a high-throughput method, it gives insight into the molecular mechanism of the transport process. It is mostly used to study the details of the ATP binding and translocation cycle of ABC transporters. However, for some ABC transporters (e.g. ABCB4) no substrate-stimulated ATPase activity can be detected. Also, ABCB4 functions as a lipid flippase [42], therefore, its function cannot be studies by vesicular transport, leaving researchers with nucleotide trapping being the only membrane-based assay to detect the interaction of compounds with the transporter. Interestingly, ABCB4 substrate drugs inhibited the amount of trapped nucleotide. The difficulty of drugs to compete with phosphatidyl choline, the abundant endogenous substrate was one of the explanations suggested by the authors [43].
Figure 3. Typical ATPase curves: Michaelis-Menten (A), "bell shaped" (B), slowly transported compound (C), inhibitor (D). The assays were performed using SB-MDR1-Sf9-ATPase membrane vesicles (Solvo Biotechnology, Budaörs, Hungary) according to a protocol based on Sarkadi et al. . Some substrates of ABC transporters stimulate the vanadate sensitive ATPase activity of membrane preparations in a standard Michaelis-Menten way (A). Other substrates are stimulators at lower concentrations, while act as inhibitors at higher concentrations resulting in a "bell shaped" curve (B). It is believed that the inhibition at higher concentrations is due to a secondary, lower affinity binding site in the extracellular part of the transporter [36]. Slowly transported compounds (C) and inhibitors (D) do not stimulate the baseline vanadate sensitive ATPase activity, but inhibit the stimulated ATPase activity measured in the presence of a strong substrate. These two types of interactions cannot be distinguished using the ATPase assays.
3.3 The nucleotide trapping assay
For ABC transporters the catalytic cycle of transport and ATP hydrolysis involves the formation of a transition state complex. This complex contains the occluded nucleotide in the nucleotide binding site, which can be stabilized with vanadate, fluoroaluminate and beryllium fluoride (with the nucleotide ADP occluded in the binding pocket). This provides the theoretical basis for a screening method, called nucleotide trapping. Vanadate is the most studied trapping agent. It has been shown for a few transporters that the amount of nucleotide trapped in the binding site by vanadate is reflective of the rate of transport, so there is an increased binding in the presence of substrates [15]. Experimentally, the isolated cell membranes are incubated with alpha-32P labeled azido-ATP for a short time (30 seconds) and after washings in the presence of cold ATP the trapped, labeled nucleotides are covalently bound to the transporter by UV photo-cross linking. The bound nucleotide is visualized by phosphorimager after the separation of proteins with SDS-PAGE [41]. Though not a high-throughput method, it gives insight into the molecular mechanism of the transport process. It is mostly used to study the details of the ATP binding and translocation cycle of ABC transporters. However, for some ABC transporters (e.g. ABCB4) no substrate-stimulated ATPase activity can be detected. Also, ABCB4 functions as a lipid flippase [42], therefore, its function cannot be studies by vesicular transport, leaving researchers with nucleotide trapping being the only membrane-based assay to detect the interaction of compounds with the transporter. Interestingly, ABCB4 substrate drugs inhibited the amount of trapped nucleotide. The difficulty of drugs to compete with phosphatidyl choline, the abundant endogenous substrate was one of the explanations suggested by the authors [43].
Membrane preparations
It is generally true that membranes can be prepared from any types of cells or tissues by any adequate method to study drug-ABC transporter interactions. There are a number of membrane preparation protocols yielding membrane preparations of different purity and physical characteristics. Also, it is important to realize the advantages and disadvantages of membranes from different sources. This section describes the most widely used membrane preparation methods and the most frequently used tissues and cell types to prepare membranes from highlighting their pros and cons in certain applications. 4.1 Membrane preparation methods The early and revolutionary work by Steck and Kant gave researches a tool to make membrane preparations containing inside-out oriented vesicles ([6], [7]). This method based on low ionic strength and divalent ions is still the most widely used method to prepare membranes from cells and tissues for ABC transporter studies. Recently, nitrogen cavitation has been successfully used to prepare membranes. This one-step spontaneous vesiculation method works in a non-hypotonic assay buffer and yields essentially equal number of vesicles of both orientations ([44], [45]). Membrane fragments generated by either of the above mentioned methods and collected by ultracentrifuging are often denoted as "partially purified" or "crude" membrane preparations. The pellet can be resuspended in a hypotonic [14] or isotonic [27] buffer and stored at -80°C or in liquid nitrogen. Most researchers use crude membrane preparations to study drug-ABC transporter interactions. Commercially available preparations are also crude membranes. ATPase assays and nucleotide trapping assays are performed using membranes suspended in hypotonic buffers ([14], [41]), while vesicular transport assays are performed both in hypotonic [23] and isotonic [27] buffers. Crude membrane preparations contain 3 types of membrane particles: lamellae, inside-out vesicles and right-side-out vesicles (figure 4). The membrane fragments originate from the plasma membrane or - to a lesser extent - from intracellular membranes [46]. Although ABC transporter proteins derived form intracellular membranes are not fully glycosylated, it has been shown in several studies that glycosylation does not modulate the biochemical characteristics of the ABC transporters ([47], [48]). It is important to note that transporters localized in membrane fragments of different orientation do not participate in all biochemical assays. In the ATPase assay and nucleotide binding assay lamellae and inside out vesicles contribute to the activity, while right-side-out vesicles do not, as the ATP binding domain of transporters is located intravesicularly. In vesicular transport experiments only the activity of transporters in inside-out vesicles is detected. There are a number of methods available based on the measurement of the activity of different enzymes located exclusively in the inner or the outer membrane leaflet in the absence or presence of various detergents (e.g. alkaline phosphatase or 5'-nucleotidase, [44]). However, membrane preparations used in publications almost never get characterized for this property. Membrane preparations can be further purified to decrease the open membrane lamellae content and increase the inside-out vesicle ratio. This is usually accomplished by sucrose gradient ultracentrifugation [46]. Immobilized concavalin A, which only binds to glycosylated extracellular domains of proteins, have also been successfully used to deplete open lamellar membranes and right side-out vesicles from crude membrane preparations ([49], [50]). Although these preparations are superior in purity they usually do not provide significantly better performance (signal-to-background ratios) in biochemical assays, therefore, the use of crude membranes is more widespread.| inside-out | open lamellar | right-side-out |
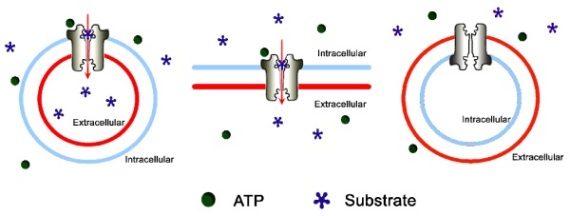 | ||
 Figure 4. The orientation of the membranes in a typical partially purified (crude) membrane preparation most frequently used in ABC transporter research. Some of the membranes are forming inside-out or right-side-out vesicles, while some of them are in an open lamellar configuration. Further purification steps can be used (e.g. sucrose gradient centrifugation) to decrease the amount of open lamellar membrane fragments and enrich inside-out vesicles.
4.2 Canalicular and basolateral liver plasma membrane vesicles
Liver plasma membrane vesicles were one of the first to be prepared and used for transport studies [51]. The successful separation of canalicular and basolateral plasma membrane vesicles [46] opened the unique opportunity to characterize the transport processes of the sinusoidal and canalicular membrane of hepatocytes separately from each other [52]. This assay system allowed the identification and characterization of an ATP-dependent bile-salt transport in canalicular rat liver plasma-membrane vesicles [28] which finally led to the discovery of the Abcb11 (Bsep, sPgp) transporter [53]. Also, canalicular plasma membrane vesicles were used to identify the bilirubin glucuronide transporter which led to the discovery of Mrp2 [54].
Liver plasma membrane vesicles serve as important tools in the discovery of transport processes and transporters of the liver; however, they are not very handy tools when it comes to large scale screening for ABC transporter interactions. The preparation of these membranes is tedious, time consuming and thereby relatively expensive; e.g. it requires multiple gradient ultraspeed centrifuging . Furthermore, the preparations contain multiple transporters disabling the identification of interactions between a drug and one specific transporter.
4.3 Membranes from mammalian cells
The first successful membrane-based ABC transporter assays were performed on membranes prepared from colchicin selected Chinese hamster ovary cells. Later, several ABC transporters were shown to transport cytotoxic compounds, and selection of cells with different chemotherapeutic agents became a standard way to acquire cell lines expressing different ABC transporters. Membranes prepared from these cells are often used in vesicular transport assay and the nucleotide trapping assay, however, the expression level of the transporter is usually not adequately high to measure the ATPase activity of the transporter. Selected cell lines often overexpress more than one transporter, whose expression profile might change with time as cells are kept in culture. The transporter involved can be identified by applying specific inhibitors. Unfortunately, specific inhibitors are not always readily available for all transporters. Zosuquidar [55] and Ko143 [56] have been successfully used to specifically inhibit ABCB1 and ABCG2, respectively, yet, no specific inhibitor is available for e.g. the members of the ABCC subfamily of transporters (MRPs) or ABCB11 (BSEP). Stable transfection of mammalian cells is another approach to run transporter specific studies. These studies are performed by comparing the transport characteristics of membranes prepared from transfected cells and the parental cell line [18].
4.4 Insect cell expression systems
The ABCB1 ATPase assay published in 1992 [14] proved that the baculoviral/Sf9 expression system is a suitable tool to study the interaction of compounds with ABC transporters. Recently, due to their improved protein expression levels researches started using High Five and Sf+ insect cells. The main advantage of the insect cell membrane preparations is the level of overexpression: as much as 3% of the total protein content of membrane preparation is the transporter of interest [14]. With only a couple of exceptions (e.g. [19]) this is the only expression system with a suitable expression of transporter to perform ATPase assays. Successful vesicular transport studies [17] and nucleotide trapping studies [41] have also been reported using ABC transporters in insect cell membranes.
Insect cell membranes are different from mammalian ones in at least two properties: (i) protein glycosylation, and (ii) the membrane lipid composition. Most outstandingly, insect membranes contain significantly less cholesterol than mammalian membranes [48]. It was shown that the substrates of ABCG2 do not stimulate the baseline ATPase activity of ABCG2 in insect membranes, while stimulation can be detected in membranes prepared from mammalian membranes [19]. Cholesterol loading and depletion experiments revealed that ABCG2-driven substrate uptake and substrate-stimulated ATPase activity are dependent on membrane cholesterol content ([48], [57]). As ABCG2 has been reported to localize into cholesterol-rich raft domains in the plasma membrane [58] cholesterol loading makes Sf9 membranes an excellent mimic of mammalian membranes. No reports indicating any functional consequence of the different glycosylation pattern of ABC transporters of insect cells have been published so far.
Altogether, the insect cell expression system is relatively inexpensive, yields high levels of protein and preparations are suitable for all types of biochemical assays used in ABC transporter research. These features have made this expression system an important tool of ABC transporter research.
4.5 Proteoliposomes
Partially or fully purified ABC transporters packed into proteoliposomes have also been used to study the biochemical characteristics of ABC transporters. Preparations have been shown to possess substrate inducible ATPase activity ([59], [60]) and ATP dependent transport [61]. This is certainly an interesting approach as these assay systems are not affected by other transporters or ATPase activities, however, the preparation of proteoliposomes is a complicated, tedious and expensive method and is unlikely to gain wide acceptance as a screening method in the pharmaceutical industry. It is also the most artificial among the systems mentioned here.
Figure 4. The orientation of the membranes in a typical partially purified (crude) membrane preparation most frequently used in ABC transporter research. Some of the membranes are forming inside-out or right-side-out vesicles, while some of them are in an open lamellar configuration. Further purification steps can be used (e.g. sucrose gradient centrifugation) to decrease the amount of open lamellar membrane fragments and enrich inside-out vesicles.
4.2 Canalicular and basolateral liver plasma membrane vesicles
Liver plasma membrane vesicles were one of the first to be prepared and used for transport studies [51]. The successful separation of canalicular and basolateral plasma membrane vesicles [46] opened the unique opportunity to characterize the transport processes of the sinusoidal and canalicular membrane of hepatocytes separately from each other [52]. This assay system allowed the identification and characterization of an ATP-dependent bile-salt transport in canalicular rat liver plasma-membrane vesicles [28] which finally led to the discovery of the Abcb11 (Bsep, sPgp) transporter [53]. Also, canalicular plasma membrane vesicles were used to identify the bilirubin glucuronide transporter which led to the discovery of Mrp2 [54].
Liver plasma membrane vesicles serve as important tools in the discovery of transport processes and transporters of the liver; however, they are not very handy tools when it comes to large scale screening for ABC transporter interactions. The preparation of these membranes is tedious, time consuming and thereby relatively expensive; e.g. it requires multiple gradient ultraspeed centrifuging . Furthermore, the preparations contain multiple transporters disabling the identification of interactions between a drug and one specific transporter.
4.3 Membranes from mammalian cells
The first successful membrane-based ABC transporter assays were performed on membranes prepared from colchicin selected Chinese hamster ovary cells. Later, several ABC transporters were shown to transport cytotoxic compounds, and selection of cells with different chemotherapeutic agents became a standard way to acquire cell lines expressing different ABC transporters. Membranes prepared from these cells are often used in vesicular transport assay and the nucleotide trapping assay, however, the expression level of the transporter is usually not adequately high to measure the ATPase activity of the transporter. Selected cell lines often overexpress more than one transporter, whose expression profile might change with time as cells are kept in culture. The transporter involved can be identified by applying specific inhibitors. Unfortunately, specific inhibitors are not always readily available for all transporters. Zosuquidar [55] and Ko143 [56] have been successfully used to specifically inhibit ABCB1 and ABCG2, respectively, yet, no specific inhibitor is available for e.g. the members of the ABCC subfamily of transporters (MRPs) or ABCB11 (BSEP). Stable transfection of mammalian cells is another approach to run transporter specific studies. These studies are performed by comparing the transport characteristics of membranes prepared from transfected cells and the parental cell line [18].
4.4 Insect cell expression systems
The ABCB1 ATPase assay published in 1992 [14] proved that the baculoviral/Sf9 expression system is a suitable tool to study the interaction of compounds with ABC transporters. Recently, due to their improved protein expression levels researches started using High Five and Sf+ insect cells. The main advantage of the insect cell membrane preparations is the level of overexpression: as much as 3% of the total protein content of membrane preparation is the transporter of interest [14]. With only a couple of exceptions (e.g. [19]) this is the only expression system with a suitable expression of transporter to perform ATPase assays. Successful vesicular transport studies [17] and nucleotide trapping studies [41] have also been reported using ABC transporters in insect cell membranes.
Insect cell membranes are different from mammalian ones in at least two properties: (i) protein glycosylation, and (ii) the membrane lipid composition. Most outstandingly, insect membranes contain significantly less cholesterol than mammalian membranes [48]. It was shown that the substrates of ABCG2 do not stimulate the baseline ATPase activity of ABCG2 in insect membranes, while stimulation can be detected in membranes prepared from mammalian membranes [19]. Cholesterol loading and depletion experiments revealed that ABCG2-driven substrate uptake and substrate-stimulated ATPase activity are dependent on membrane cholesterol content ([48], [57]). As ABCG2 has been reported to localize into cholesterol-rich raft domains in the plasma membrane [58] cholesterol loading makes Sf9 membranes an excellent mimic of mammalian membranes. No reports indicating any functional consequence of the different glycosylation pattern of ABC transporters of insect cells have been published so far.
Altogether, the insect cell expression system is relatively inexpensive, yields high levels of protein and preparations are suitable for all types of biochemical assays used in ABC transporter research. These features have made this expression system an important tool of ABC transporter research.
4.5 Proteoliposomes
Partially or fully purified ABC transporters packed into proteoliposomes have also been used to study the biochemical characteristics of ABC transporters. Preparations have been shown to possess substrate inducible ATPase activity ([59], [60]) and ATP dependent transport [61]. This is certainly an interesting approach as these assay systems are not affected by other transporters or ATPase activities, however, the preparation of proteoliposomes is a complicated, tedious and expensive method and is unlikely to gain wide acceptance as a screening method in the pharmaceutical industry. It is also the most artificial among the systems mentioned here.
Correlations of membrane test data with data from other in vitro
A number of validation studies indicated discrepancies between results obtained using the ATPase assay and the results of cellular ABCB1 assays like monolayer efflux assays utilizing Caco-2 or MDCKII-MDR1 cells or the Calcein assay (for further information on the Calcein assay pls. refer to [62]). On one hand, except for compounds that exhibited very low passive permeability, the ATPase assay and the Calcein assay showed good correlations. On the other hand, highly permeable compounds showed activation in the ATPase assay and were not shown to be transported in the monolayer efflux assay [63]. Authors of the above paper suggested a nomenclature where compounds are classified as "transported substrates" if the transport of the compound is detected in the monolayer efflux assay, while compounds that show interactions in the ATPase assay and the Calcein assay, but do not appear to be transported in the monolayer efflux assay are classified as "nontransported substrates". As the explanation for the discrepancies is probably not related to the interaction of the compound with the transporter, this nomenclature is highly misleading. Therefore, it is important to emphasize that "nontransported substrates" do interact with the transporter and get transported (translocated through the lipid bilayer in an ATP dependent manner). Several of them were shown to inhibit the transport of digoxin in the monolayer efflux assay suggesting that these compounds might cause clinical drug-drug interactions [64]. Furthermore, it was shown that ABCB1 does contribute to a limited brain-to-plasma ratio for some of these compounds (e.g. verapamil [65] and ketoconazole [66]) indicating that these compounds are, indeed, transported in vivo. In conclusion, monolayer efflux assays do not identify some clinically relevant interactions of highly permeably substrates with transporters, providing false negative results, while these interactions are readily detected by the ATPase assay. It is often proposed that the ATPase assay indicates the interaction of a test compound with the transporter at significantly higher concentrations than the concentration ever reached in vivo. This is often interpreted as an interaction "not relevant". It is important to emphasize that the fact that the amount of inorganic phosphate liberated by the transporter at lower substrate concentrations cannot be detected does not mean that the transporter is inactive at those concentrations. A typical example of this situation is the transport of prazosine by ABCG2. The rate of ATP cleavage can only be detected above 10 µM in the ATPase assay [19]. However, the transport of 3H-prazosine in a monolayer efflux assay can be detected at concentrations as low as 100 nM [67]. This indicates that interactions detected in the ATPase assay at high concentrations are also relevant at lower concentrations. The ATPase assay allows the determination of the affinity of the compound to the transporter and the amount of inorganic phosphate generated by the transporter at a certain concentration of the test compound. These parameters are often correlated to readouts such as distribution of the compounds across barriers (e.g. monolayer studies [63], or brain-to-plasma ratios [68]). It is important to emphasize that the ATPase assay does not give us precise information on the transport rate, and by no means gives us information on the magnitude of the transport in relation to the passive permeability of the compounds. Therefore, it is in principle impossible to find any correlation between the results of the ATPase assay - especially affinity type of parameters - with in vitro or in vivo distribution figures. In sum, while the ATPase assay is an excellent tool to detect the interactions of test compounds with transporters the results of this assay alone cannot be used to predict how the interaction is going to modulate the distribution of the compound at a barrier containing the transporter. Our current understanding of how membrane based assays correlate with each other and with the results of other assays is primarily based on studies conducted on the ABCB1 transporter. Most substrates of ABCB1 are highly permeable thereby making direct monolayer and vesicular transport assays impossible to perform. Running correlation studies on other transporters with different substrate profile might help us to overcome this problem. Several substrates of the ABCC family and ABCG2 are ionic compounds with very low passive permeability. It was shown for ABCG2 ATPase and vesicular transport using estrone-3-sulfate as substrate that kinetic data obtained are comparable [19]. Further studies using low permeability compounds will be necessary to show that this correlation is universally true. Also, this indicates that we have to be very careful in drawing general conclusions on transporter assays based on observations made using ABCB1 assays.Conclusion
In the last 15 years our understanding of how ABC transporters are involved in the modulation of the pharmacokinetic properties of drugs established a new area of interest in the discovery and development of new pharmaceuticals. Assays based on different membrane preparations were one of the first methods invented and they turned out to be valuable tools to gain deeper insight into the nature of drug-ABC transporter interactions. Membranes prepared from different sources have been used and characterized, and based on the biochemical characteristics of the transport process a number of different assay types have been invented. These assays - further developed and refined - are now available for pharmaceutical industry to be incorporated in the standard pipeline of drug development. The ATPase assay is very helpful in detecting the interaction of test compounds with the transporter. In case stimulation of the transporter ATPase activity is detected we can conclude that the compound is probably a transported substrate. It is the only in vitro tool that readily predicts the substrate nature of highly permeable drugs. However, kinetic data acquired is not easily interpreted and correlated to other assays. For compounds that only show inhibition in the ATPase assay interpretation of the data is even further limited: we cannot tell if the compound is an inhibitor or a slowly transported substrate of the transporter. Direct vesicular transport is an excellent tool to detect the actual transport and analyze transport kinetics in detail, yet, only compounds with low passive permeability can be detected using this method. The indirect vesicular transport assay indicates the interaction regardless of the passive permeability of the test compound, however, being an indirect assay, will not indicate the nature (substrate/inhibitor) of the interaction. This type of assay is a perfect tool to screen for ABC transporter mediated drug-drug or drug-endogenous substrate interactions. The rational use of the nucleotide trapping assay looks very limited for pharmaceutical industry at the moment.Expert opinion
The area of transporter research is at its full height. New transporters, new drug-transporter interactions are daily discovered. The pharmacological relevance of some of these interactions also drew interest of researchers working in the field of drug discovery. It is not hard to perceive that some assays for some of the transporters will be incorporated in the pharmaceutical pipeline as standard assays of drug discovery and development. Assay selection should be based on (i) the question addressed (drug - transporter interaction, mechanism of interaction (substrate vs inhibitor), drug - drug interaction, drug - endobiotics interaction) and (ii) the passive permeability of the compound (table 2.)| Assay | Assay mode | Permeability space | Interaction detected |
| ATPase assay | Activation | From high to low | Indicates high turnover transport |
| Inhibition | From high to low | Detects slowly transported compounds and inhibitors | |
| Vesicular transport assay | Direct | Very low | Direct transport measurement, suitable for determination of kinetic data |
| Indirect | From high to low | Detects compounds that interact with the transporter, drug-drug interaction models | |
| Nucleotide trapping assay | From high to low | Modulation of ATP binding and translocation cycle of the transporter |
Annotated bibliography
1. GLAVINAS H, KRAJCSI P, CSEREPES J, et al., The role of ABC transporters in drug resistance, metabolism and toxicity. Curr Drug Deliv (2004) 1(1):27-42.2. SARKADI B, HOMOLYA L, SZAKACS G, et al., Human multidrug resistance ABCB and ABCG transporters: participation in a chemoimmunity defense system. Physiol Rev (2006) 86(4):1179-236.
Comprehensive review of the ABCB1 and ABCG2 proteins.
3. MIZUNO N, NIWA T, YOTSUMOTO Y, et al., Impact of drug transporter studies on drug discovery and development. Pharmacol Rev (2003) 55(3):425-61.
4. LIN JH, YAMAZAKI M, Role of P-glycoprotein in pharmacokinetics: clinical implications. Clin Pharmacokinet (2003) 42(1):59-98.
5. MARTONOSI A, FERETOS R, Sarcoplasmic Reticulum. I. The Uptake of Ca++ by Sarcoplasmic Reticulum Fragments. J Biol Chem (1964) 239(648-58.)
6. STECK TL, WEINSTEIN RS, STRAUS JH, et al., Inside-out red cell membrane vesicles: preparation and purification. Science (1970) 168(928):255-7.
The method of inside-out vesicle preparation by the hypotonic lysis method published in this paper is the basis for most of the protocols used for membrane preparation in modern ABC transporter research.
7. STECK TL, KANT JA, Preparation of impermeable ghosts and inside-out vesicles from human erythrocyte membranes. Methods Enzymol (1974) 31(Pt A):172-80.
8. JULIANO RL, LING V, A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta (1976) 455(1):152-62.
9. SHEN DW, FOJO A, CHIN JE, et al., Human multidrug-resistant cell lines: increased mdr1 expression can precede gene amplification. Science (1986) 232(4750):643-5.
10. CHEN CJ, CHIN JE, UEDA K, et al., Internal duplication and homology with bacterial transport proteins in the mdr1 (P-glycoprotein) gene from multidrugresistant human cells. Cell (1986) 47(3):381-9. 11. UEDA K, CORNWELL MM, GOTTESMAN MM, et al., The mdr1 gene, responsible for multidrug-resistance, codes for P-glycoprotein. Biochem Biophys Res Commun (1986) 141(3):956-62.
12. HORIO M, GOTTESMAN MM, PASTAN I, ATP-dependent transport of vinblastine in vesicles from human multidrug-resistant cells. Proc Natl Acad Sci U S A (1988) 85(10):3580-4.
The first paper describing the ABCB1 vesicular transport assay.
13. DOIGE CA, SHAROM FJ, Transport properties of P-glycoprotein in plasma membrane vesicles from multidrug-resistant Chinese hamster ovary cells. Biochim Biophys Acta (1992) 1109(2):161-71.
14. SARKADI B, PRICE EM, BOUCHER RC, et al., Expression of the human multidrug resistance cDNA in insect cells generates a high activity drugstimulated membrane ATPase. J Biol Chem (1992) 267(7):4854-8.
The first paper describing the ABCB1 ATPase assay.
15. URBATSCH IL, SANKARAN B, WEBER J, et al., P-glycoprotein is stably inhibited by vanadate-induced trapping of nucleotide at a single catalytic site. J Biol Chem (1995) 270(33):19383-90.
The first paper describing the ABCB1 nucleotide trapping assay.
16. ABC transporters: biochemical, cellular and molecular aspects. Methods Enzymol (1998) 292
This volume of Methods in enzymology is dedicated to methodologies used to study ABC transporters.
17. BAKOS E, EVERS R, SZAKACS G, et al., Functional multidrug resistance protein (MRP1) lacking the N-terminal transmembrane domain. J Biol Chem (1998) 273(48):32167-75.
18. ZENG H, LIU G, REA PA, et al., Transport of amphipathic anions by human multidrug resistance protein 3. Cancer Res (2000) 60(17):4779-84.
19. GLAVINAS H, KIS E, PAL A, et al., ABCG2 (BCRP/MXR) ATPase assay - a useful tool to detect drug - transporter interactions. Drug Metab Dispos (2007)
20. SHAROM FJ, YU X, LU P, et al., Interaction of the P-glycoprotein multidrug transporter (MDR1) with high affinity peptide chemosensitizers in isolated membranes, reconstituted systems, and intact cells. Biochem Pharmacol (1999) 58(4):571-86.
Paper covering multiple important in vitro methods to study drug - transporter interactions.
21. PAULUSMA CC, BOSMA PJ, ZAMAN GJ, et al., Congenital jaundice in rats with a mutation in a multidrug resistance-associated protein gene. Science (1996) 271(5252):1126-8.
22. ZELCER N, HUISMAN MT, REID G, et al., Evidence for two interacting ligand binding sites in human multidrug resistance protein 2 (ATP binding cassette C2). J Biol Chem (2003) 278(26):23538-44.
23. BODO A, BAKOS E, SZERI F, et al., Differential modulation of the human liver conjugate transporters MRP2 and MRP3 by bile acids and organic anions. J Biol Chem (2003) 278(26):23529-37.
These two papers provide excellent kinetic analysis of drug interactions on the human MRP2 transporter.
24. CUI Y, KONIG J, KEPPLER D, Vectorial transport by double-transfected cells expressing the human uptake transporter SLC21A8 and the apical export pump ABCC2. Mol Pharmacol (2001) 60(5):934-43.
25. SASAKI M, SUZUKI H, ITO K, et al., Transcellular transport of organic anions across a double-transfected Madin-Darby canine kidney II cell monolayer expressing both human organic anion-transporting polypeptide (OATP2/SLC21A6) and Multidrug resistance-associated protein 2 (MRP2/ABCC2). J Biol Chem (2002) 277(8):6497-503.
26. BARTHOLOME K, RIUS M, LETSCHERT K, et al., Data-based mathematical modeling of vectorial transport across double-transfected polarized cells. Drug Metab Dispos (2007) 35(9):1476-81.
27. BARTHOLOME K, RIUS M, LETSCHERT K, et al., Data-based mathematical modeling of vectorial transport across double-transfected polarized cells. Drug Metab Dispos (2007) 35(9):1476-81.
28. STIEGER B, O'NEILL B, MEIER PJ, ATP-dependent bile-salt transport in canalicular rat liver plasma-membrane vesicles. Biochem J (1992) 284 ( Pt 1)(67-74).
29. STRAUTNIEKS SS, BULL LN, KNISELY AS, et al., A gene encoding a liverspecific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nat Genet (1998) 20(3):233-8.
30. BAKOS E, EVERS R, SINKO E, et al., Interactions of the human multidrug resistance proteins MRP1 and MRP2 with organic anions. Mol Pharmacol (2000) 57(4):760-8.
31. LESPINE A, DUPUY J, ORLOWSKI S, et al., Interaction of ivermectin with multidrug resistance proteins (MRP1, 2 and 3). Chem Biol Interact (2006) 159(3):169-79.
32. WILLIAMSON G, AEBERLI I, MIGUET L, et al., Interaction of positional isomers of quercetin glucuronides with the transporter ABCC2 (cMOAT, MRP2). Drug Metab Dispos (2007) 35(8):1262-8.
33. ZHANG L, LIN G, KOVACS B, et al., Mechanistic study on the intestinal absorption and disposition of baicalein. Eur J Pharm Sci (2007) 31(3-4):221-31.
34. CURRIER SJ, UEDA K, WILLINGHAM MC, et al., Deletion and insertion mutants of the multidrug transporter. J Biol Chem (1989) 264(24):14376-81.
35. BROXTERMAN HJ, PINEDO HM, KUIPER CM, et al., Induction by verapamil of a rapid increase in ATP consumption in multidrug-resistant tumor cells. Faseb J (1988) 2(7):2278-82.
36. LITMAN T, NIELSEN D, SKOVSGAARD T, et al., ATPase activity of Pglycoprotein related to emergence of drug resistance in Ehrlich ascites tumor cell lines. Biochim Biophys Acta (1997) 1361(2):147-58.
37. BUXBAUM E, Co-operative binding sites for transported substrates in the multiple drug resistance transporter Mdr1. Eur J Biochem (1999) 265(1):64-70.
38. AL-SHAWI MK, POLAR MK, OMOTE H, et al., Transition state analysis of the coupling of drug transport to ATP hydrolysis by P-glycoprotein. J Biol Chem (2003) 278(52):52629-40.
Good paper on transport and ATPase coupling of ABCB1.
39. SHIRASAKA Y, ONISHI Y, SAKURAI A, et al., Evaluation of human Pglycoprotein (MDR1/ABCB1) ATPase activity assay method by comparing with in vitro transport measurements: Michaelis-Menten kinetic analysis to estimate the affinity of P-glycoprotein to drugs. Biol Pharm Bull (2006) 29(12):2465-71.
40. GOLDBERG H, LING V, WONG PY, et al., Reduced cyclosporin accumulation in multidrug-resistant cells. Biochem Biophys Res Commun (1988) 152(2):552-8.
41. SZABO K, WELKER E, BAKOS, et al., Drug-stimulated nucleotide trapping in the human multidrug transporter MDR1. Cooperation of the nucleotide binding domains. J Biol Chem (1998) 273(17):10132-8.
42. VAN HELVOORT A, SMITH AJ, SPRONG H, et al., MDR1 P-glycoprotein is a lipid translocase of broad specificity, while MDR3 P-glycoprotein specifically translocates phosphatidylcholine. Cell (1996) 87(3):507-17.
43. SMITH AJ, VAN HELVOORT A, VAN MEER G, et al., MDR3 P-glycoprotein, a phosphatidylcholine translocase, transports several cytotoxic drugs and directly interacts with drugs as judged by interference with nucleotide trapping. J Biol Chem (2000) 275(31):23530-9.
44. LOE DW, ALMQUIST KC, DEELEY RG, et al., Multidrug resistance protein (MRP)-mediated transport of leukotriene C4 and chemotherapeutic agents in membrane vesicles. Demonstration of glutathione-dependent vincristine transport. J Biol Chem (1996) 271(16):9675-82.
45. VOLK EL, SCHNEIDER E, Wild-type breast cancer resistance protein (BCRP/ABCG2) is a methotrexate polyglutamate transporter. Cancer Res (2003) 63(17):5538-43.
46. MEIER PJ, SZTUL ES, REUBEN A, et al., Structural and functional polarity of canalicular and basolateral plasma membrane vesicles isolated in high yield from rat liver. J Cell Biol (1984) 98(3):991-1000.
47. GERMANN UA, CHAMBERS TC, AMBUDKAR SV, et al., Characterization of phosphorylation-defective mutants of human P-glycoprotein expressed in mammalian cells. J Biol Chem (1996) 271(3):1708-16.
48. PAL A, MEHN D, MOLNAR E, et al., Cholesterol potentiates ABCG2 activity in a heterologous expression system: improved in vitro model to study function of human ABCG2. J Pharmacol Exp Ther (2007) 321(3):1085-94.
49. KONDO T, DALE GL, BEUTLER E, Simple and rapid purification of inside-out vesicles from human erythrocytes. Biochim Biophys Acta (1980) 602(1):127-30.
50. KONDO T, DALE GL, BEUTLER E, Simple and rapid purification of inside-out vesicles from human erythrocytes. Biochim Biophys Acta (1980) 602(1):127-30.
51. BOYER JL, ALLEN RM, NG OC, Biochemical separation of Na+,K+-ATPase from a "purified" light density, "canalicular"-enriched plasma membrane fraction from rat liver. Hepatology (1983) 3(1):18-28.
52. MEIER PJ, ST MEIER-ABT A, BARRETT C, et al., Mechanisms of taurocholate transport in canalicular and basolateral rat liver plasma membrane vesicles. Evidence for an electrogenic canalicular organic anion carrier. J Biol Chem (1984) 259(16):10614-22.
53. GERLOFF T, STIEGER B, HAGENBUCH B, et al., The sister of P-glycoprotein represents the canalicular bile salt export pump of mammalian liver. J Biol Chem (1998) 273(16):10046-50.
54. JEDLITSCHKY G, LEIER I, BUCHHOLZ U, et al., ATP-dependent transport of bilirubin glucuronides by the multidrug resistance protein MRP1 and its hepatocyte canalicular isoform MRP2. Biochem J (1997) 327 ( Pt 1)(305-10.
Good paper on MRP2/Mrp2 and the methods to study transporter function.
55. SLATE DL, BRUNO NA, CASEY SM, et al., RS-33295-198: a novel, potent modulator of P-glycoprotein-mediated multidrug resistance. Anticancer Res (1995) 15(3):811-4.
56. ALLEN JD, VAN LOEVEZIJN A, LAKHAI JM, et al., Potent and specific inhibition of the breast cancer resistance protein multidrug transporter in vitro and in mouse intestine by a novel analogue of fumitremorgin C. Mol Cancer Ther (2002) 1(6):417-25.
57. TELBISZ A, MULLER M, OZVEGY-LACZKA C, et al., Membrane cholesterol selectively modulates the activity of the human ABCG2 multidrug transporter. Biochim Biophys Acta (2007) 1768(11):2698-713.
58. STORCH CH, EHEHALT R, HAEFELI WE, et al., Localization of the human breast cancer resistance protein (BCRP/ABCG2) in lipid rafts/caveolae and modulation of its activity by cholesterol in vitro. J Pharmacol Exp Ther (2007) 323(1):257-64.
59. DOIGE CA, YU X, SHAROM FJ, ATPase activity of partially purified Pglycoprotein from multidrug-resistant Chinese hamster ovary cells. Biochim Biophys Acta (1992) 1109(2):149-60.
60. AMBUDKAR SV, LELONG IH, ZHANG J, et al., Partial purification and reconstitution of the human multidrug-resistance pump: characterization of the drug-stimulatable ATP hydrolysis. Proc Natl Acad Sci U S A (1992) 89(18):8472-6.
Another good paper on ABCB1 transport cycle.
61. SHAROM FJ, YU X, DOIGE CA, Functional reconstitution of drug transport and ATPase activity in proteoliposomes containing partially purified P-glycoprotein. J Biol Chem (1993) 268(32):24197-202.
62. HOMOLYA L, HOLLO M, MULLER M, et al., A new method for a quantitative assessment of P-glycoprotein-related multidrug resistance in tumour cells. Br J Cancer (1996) 73(7):849-55.
63. POLLI JW, WRING SA, HUMPHREYS JE, et al., Rational use of in vitro Pglycoprotein assays in drug discovery. J Pharmacol Exp Ther (2001) 299(2):620-8.
Basic paper on the effect of passive permeability on assay reliability.
64. RAUTIO J, HUMPHREYS JE, WEBSTER LO, et al., In vitro p-glycoprotein inhibition assays for assessment of clinical drug interaction potential of new drug candidates: a recommendation for probe substrates. Drug Metab Dispos (2006) 34(5):786-92.
The paper shows that assay selection is crucial. There is limited correlation between bidirectional transport assay in MDCKIIMDR1 cells and the drugdrug interaction potential of the same compound assayed in the same cell line using digoxin as reporter compound.
65. The paper shows that assay selection is crucial. There is limited correlation between bidirectional transport assay in MDCKIIMDR1 cells and the drugdrug interaction potential of the same compound assayed in the same cell line using digoxin as reporter compound.
66. FAHEY JM, PRITCHARD GA, MOLTKE LL, et al., Effects of ketoconazole on triazolam pharmacokinetics, pharmacodynamics and benzodiazepine receptor binding in mice. J Pharmacol Exp Ther (1998) 285(1):271-6.
67. XIAO Y, DAVIDSON R, SMITH A, et al., A 96-well efflux assay to identify ABCG2 substrates using a stably transfected MDCK II cell line. Mol Pharm (2006) 3(1):45-54.
68. XIAO Y, DAVIDSON R, SMITH A, et al., A 96-well efflux assay to identify ABCG2 substrates using a stably transfected MDCK II cell line. Mol Pharm (2006) 3(1):45-54.
69. WU CY, BENET LZ, Predicting drug disposition via application of BCS: transport/absorption/ elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm Res (2005) 22(1):11-23.
Informative review on the effect of transporters on pharmacokinetics of drugs.
70. NOE J, STIEGER B, MEIER PJ, Functional expression of the canalicular bile salt export pump of human liver. Gastroenterology (2002) 123(5):1659-66.
